
Natural selection has immense and important effect on natural populations and is crucial in determining the fate of these populations in the context of rapid contemporary and human-mediated habitat changes. We combine evolutionary, ecological, and “omics” approaches to study the role of natural selection in driving evolution of several taxa in the wild. We highlight important areas of our research below.
Genomic consequences of hybridization
“There was never a single trunk population in the human past. It has been mixtures all the way down.” – David Reich, Who We Are and How We Got Here: Ancient DNA and the New Science of the Human Past
Admixture, the genetic merging of parental populations resulting in mixed ancestry, occurs frequently in nature, creating naturally admixed populations. For example, numerous admixture events have occurred between human populations across the world, which have shaped genetic ancestry in modern humans. This process often creates populations which are mosaics of different ancestries. Admixture events can be caused by range expansions and colonization of novel environments. Admixed genomes offer novel insights about the evolutionary history of natural populations and the role of selection in shaping genomes in the face gene flow and possibilities of speciation. While studies of admixed human genomes have made considerable progress, non-model organisms have been less explored to understand these processes. We combine extensive spatial, temporal, and species/hybrid level sampling of thousands of individuals to study the genomic outcomes of admixture and hybridization and how these genomes are shaped by selection. For instance, we empirically demonstrated that patterns of introgression in a contemporary hybrid zone in Lycaeides butterflies predict patterns of ancestry in geographically adjacent, older hybrid populations. We are now leveraging genomic sampling of thousands of samples of several species and hybrid populations to generate a map of archaic ancestry and parse the role admixture versus incomplete lineage sorting in shaping ancestry across admixed genomes. Our work provides a novel approach to study a basic question about admixed mosaic genomes which has only been applied to humans and primates but can be extended to non-model organisms.
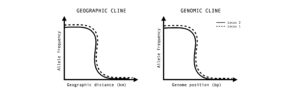
Figure 1: An illustration of clines. Y-axes track allele frequency of a given locus. X-axes correspond to position along geographic transects (ecological gradient) or hybrid indexes (genomic gradient) in the case of genomic cline analyses.
Genomic basis of adaptive evolution
“Listen, if there’s one thing the history of evolution has taught us is that life will not be contained. Life breaks free. It expands to new territories, and it crashes through barriers painfully, maybe even dangerously, but . . . life finds a way”. – Jurassic Park
The amount and nature of genetic variation available to natural selection affect the rate, course and outcome of evolution. Consequently, the study of the genetic basis of adaptive evolutionary change has occupied biologists for decades, but progress has been hampered by the lack of resolution and the absence of a genome-level perspective. Technological advances in recent years should now allow us to answer many long-standing questions about the nature of adaptation. One question which arises in the study of genomic basis of adaptation, is whether rapid adaptation occurs due to standing genetic variation or by accumulating de novo mutations. We study the genomic basis of local adaptation to novel environments and contemporary habitat changes. For example, we have demonstrated repeated instances of adaptation to novel host plants in herbivorous insects and parallel adaptation to crucial climate variables across several species of Timema stick insects. We are now using widespread geographic sampling along longitudinal, latitudinal, and climatic gradients of butterflies and forest trees to understand the genetic architecture of local adaptation and predict species response to future environmental changes. Our work provides a comprehensive understanding of the genetics of adaptation in the face of contemporary and human-induced habitat changes.
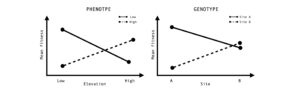
Figure 2: In spatially heterogeneous landscapes, divergent selection in contrasting environments can result in the evolution of local adaptation. Local adaptation can be measured by quantifying mean fitness of an individual at a phenotypic level or genotypic level. We would expect that a local phenotype or genotype will provide higher fitness in the local environment compared to the non-local environment creating an antagonistic pattern.
Genomic signatures of co-evolutionary interactions
“Now, here, you see, it takes all the running you can do, to keep in the same place. If you want to get somewhere else, you must run at least twice as fast as that!” – Lewis Carroll, Alice in Wonderland and Through the Looking Glass
Inter-specific coevolution is ubiquitous and shapes genetic and phenotypic diversity of interacting species. For example, co-evolution between herbivorous insects and their host plants is one of the important drivers in the great diversity of both these lineages. Except for a few important model systems, the process of co-evolution itself has rarely been studied owing to a lack of tools—instead, researchers have focused on macroevolutionary patterns that might have been created by co-evolution. Furthermore, understanding of the proximate mechanisms which underlie co-evolutionary interactions is limited. Recent advances in high through-put sequencing technologies provide a novel opportunity to dissect the genetic, dynamics, and consequences of co-evolution over space and time. For example, we have demonstrated that herbivorous insects show genomic signatures of adaptation to novel host plants and genomic regions associated with specific aspects of host plant use can be under selection. We are now using the iconic and endemic species pair of B. philenor hirsuta butterflies, and its host plant, A. californica Torr. to study their co-evolution at the genomic, spatial, and temporal scale. Our work provides a novel combination of proximate and ultimate approaches to study co-evolution in nature in a non-model organisms which are impacted by rapid climate change.
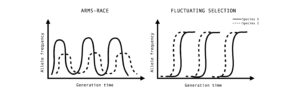
Figure 3: Co-evolutionary dynamics observed for a given pair of species. Allele frequency dynamics coevolution can show an escalating arms race dynamics due to selective sweeps occurring from standing genetic variation and balancing selection or red queen dynamics due to novel mutations and negative frequency-dependent selection.
Quantifying predictability of evolution
“Wind back the tape of life to the early days of the Burgess Shale; let it play again from an identical starting point, and the chance becomes vanishingly small that anything like human intelligence would grace the replay.” – Stephen Jay Gould, Wonderful Life: The Burgess Shale and the Nature of History
Predictability in science is crucial to test robustness of experimental and to forecast or retrospect the fate of evolving systems. In evolutionary biology, natural selection can drive predicable outcomes at several scales (space, time), levels (genotypic, phenotypic), for traits (physiology, behavior), and in forms (repeated, mechanistic). However, we can be limited in our predictability of evolution due to random evolutionary processes which limit predictability (such as drift or mutations) or due to the lack of data. Moreover, a binary outcome of prediction is limiting and a more comprehensive quantitative framework is required to study and understand predictability of evolution. Our work has an overarching goal to quantify predictability in different areas of evolutionary biology. We are expanding our previous work on predictability by testing for repeated instances of specialization in herbivorous insects, convergent mechanisms of toxin sequestration, parallel adaptation to climate, and identifying repeatable occurrence of traits on spatial and temporal scales. In the grander scheme of things, we hope to use a comparative framework which can advance our knowledge of predictability of evolution.
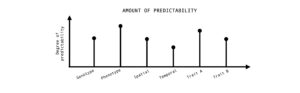
Figure 4: Predictability of evolution can be quantified at different levels (genotype or phenotype), scales (spatial or temporal), biological traits (life-history or physiology). There are various factors which can influence the degree of predictability and a more comprehensive understanding can be gained by a quantitative and comparative framework.